RNA, DNA and Protein Structure Viewer
RNA/DNA secondary structure fold viewer
By default this viewer is only shown when an oligo sequence is selected. If you wish to use RNA fold on a non-oligo sequence, go to Tools → Preferences → Appearance and Behavior and enable the option Show DNA/RNA fold view on all sequence. This will show the tab for any sequence less than 3000 bp. If the selected sequence is DNA, the tab will be labelled DNA Fold and if it is RNA it will be labelled RNA Fold.
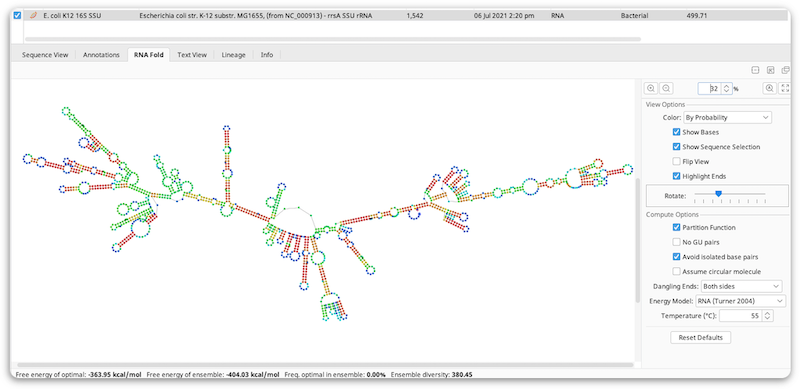
The fold prediction is performed by the Vienna package RNAfold tool. Information on the options for this tool can be found at the following web page: http://www.tbi.univie.ac.at/~ivo/RNA/RNAfold.html.
The View Options allow you to turn off/on and color the bases, flip the coordinates, highlight the start (blue) and end (red) of the sequence and rotate the model. As with other viewers, you can zoom in on the model and drag the view around, or use the scrollwheel using the same keyboard modifiers as the sequence viewer. Selection is synchronized between the sequence view and the fold view. In addition, when in split view mode, the fold viewer will scroll to the selected area when zoomed in.
By default, Color by probability is used where red bases are the ones with the strongest probability of the bases being paired with each other in paired regions, or being unpaired in unpaired regions. Green is the middle ground and blue is the lowest probability. Color by probability is only available when using the Partition Function.
Compute Options will rerun RNAfold when you change their settings, so depending on the size of the sequence there may be a noticeable recompute time.
3D protein structure viewer
For molecular structure documents, such as PDB documents, this displays an interactive three dimensional view of the structure.
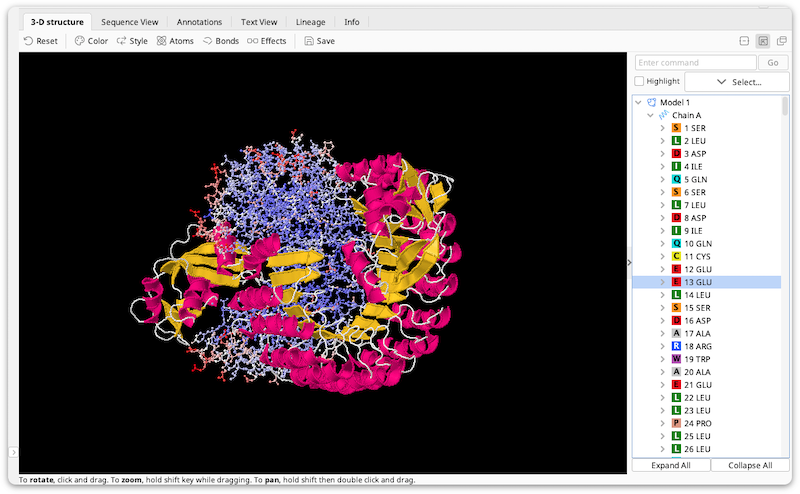
Structure View Manipulation
-
Click and drag the mouse to rotate the structure.
-
Hold the Alt or Shift key then click and drag to zoom in/out
-
Hold the Ctrl key then right-click and drag to pan, or, if you are using a Mac, click and hold, press Ctrl and Alt/Option then drag to pan.
Selection Controls
To the right of the structure are controls that let you control the selected part of the structure.
-
If the structure you are viewing contains more than one model, the model combo box will you choose between them.
-
The select button lets you select all, none or the nonselected region of the structure, as well as by element, group type or secondary structure.
-
The highlight selected checkbox lets you select whether to highlight the selected atoms in the structure view.
-
The structure tree shows the atoms in the structure in a tree format. Click on regions in the tree to select those regions. You can also Shift-click and Ctrl-click to select multiple regions at once.
-
The command box lets you type in arbitrary jmol scripting commands. To see some examples, select one of the pre-populated options in the box's drop-down. For a complete description of the commands you can use, see http://www.stolaf.edu/academics/chemapps/jmol/docs.
Display Menu
At the top of the viewer is the display menu. Here you can modify the appearance of the structure.
-
Reset lets you reset the position of the structure, reset the appearance of the structure to the default, or reset the appearance of the structure to its appearance when it was last saved.
-
Color lets you change the color scheme of the selected region of the atom.
-
Style lets you change the style of the selected region of the molecule eg to spacefill or cartoon view.
-
Atoms lets you hide atoms or change their size in the selected region of the molecule. You can also choose whether to show hydrogen atoms and atom symbols.
-
Bonds lets you hide bonds or change their size in the selected region of the molecule. Covalent/ionic bonds, hydrogen bonds and disulfide bonds can be affected separately.
-
Effects lets you toggle spin, antialiasing, stereo and slabbing effects for the whole molecule.
-
Save saves the current appearance of the molecule.